


Incógnitas de nuestro ADN
María Emilia Beyer

Shutterstock
Apenas entre el 1 y el 2% de nuestro material genético está compuesto por segmentos de ADN que tienen genes con los códigos necesarios para fabricar las proteínas que nos constituyen y nos permiten funcionar. El resto, se pensaba, es inservible: basura. Pero varias investigaciones han encontrado que no necesariamente es así y que es posible que nos aguarden muchas sorpresas.
Un inglés alto, flaco y desgarbado irrumpe en el bar Eagle en Cambridge, Inglaterra, para dar una noticia estruendosa y poco humilde: “hemos descubierto el secreto de la vida”. El año es 1953. El personaje es Francis Crick, quien acompañado por James Watson, acaba de anunciar el descubrimiento de la estructura en forma de doble hélice que tiene el ácido desoxirribonucleico, o ADN. Apenas dos meses después, la revista Nature publicó el artículo sobre el hallazgo que, con escasas 120 líneas, revolucionó la ciencia.
Watson y Crick no estaban equivocados en su aseveración inicial, pues para comprender el funcionamiento de la vida, es fundamental conocer cómo es el ADN. Pero los hallazgos estaban por comenzar. El Proyecto Genoma Humano que fue anunciado en el 2001 (véase ¿Cómo ves? No. 37 y No. 146), y la posterior lectura del genoma de otras especies en las últimas décadas nos han llevado de sorpresa en sorpresa, y de paso nos han brindado un par de moralejas biológicas.
En 2001 se estimaba que el genoma humano constaba de unos 100 000 genes, pero a medida que mejoraron las técnicas para identificar las secciones del genoma humano que tienen genes que codifican proteínas esta cifra cayó en picada. Hoy se estima que el número de genes para la especie humana apenas rebasa los 20 000.
Fantasías genómicas
Uno de los hallazgos más sorprendentes de la lectura o desciframiento de nuestra receta bioquímica es la escasez de genes. Hacia el 2001 se estimaba que el genoma humano constaba de unos 100 000 genes. Esta cifra surgió del paradigma “un gen, una proteína”, que consideraba que si los genes son códigos bioquímicos con instrucciones precisas para fabricar proteínas, y si los seres humanos tenemos aproximadamente 100 000 proteínas diferentes, entonces habría una correspondencia directa entre el número total de ambos elementos. Por eso resultó muy sorprendente encontrar en un conteo inicial que los seres humanos teníamos unos 30 000 genes.
Este número, sin embargo, estaba errado. La caída libre en el conteo continuó a medida que mejoraron las técnicas para identificar las secciones del genoma humano que tienen genes que codifican proteínas, y así, actualmente se estima que el número de genes para la especie humana apenas rebasa los 20 000. Este número es muy inferior al que se esperaba inicialmente, pero ¿por qué resultó tan asombroso? Si consideramos que los seres humanos somos los animales con mayor impacto en el planeta, capaces de reflexionar sobre nosotros mismos, construir arte y cultura o viajar al espacio, se podría soñar con una composición genómica inmensa y llena de complejidades, pero resulta que somos bastante parecidos al resto de los seres vivos. No parece que tengamos nada espectacular en nuestra receta bioquímica.
Se ha encontrado que el ADN de ciertos peces pulmonados es 40 veces más grande que el nuestro. Por su parte, la planta del maíz nos rebasa por varios miles de genes, y la mosca de la fruta tiene apenas 6 000 genes menos que nosotros. La diferencia en el conteo genómico entre el gusano Caenorhabditis elegans (de apenas 1 mm de largo) y nosotros es de unos cuantos cientos de genes.
La incredulidad inicial ante estas cifras no provino exclusivamente de la soberbia evolutiva que nos acompaña como especie. También se conocía la longitud de nuestra cadena de ADN, compuesta por 3 000 millones de nucleótidos. Haciendo cuentas, esta longitud de material genético daba un filamento tan largo, que dentro de una sola célula podíamos guardar aproximadamente dos metros de este tesoro biológico. Y si sumamos esos dos metros de ADN por cada célula del cuerpo humano, alcanzamos una longitud de cadena apabullante, que permitiría a la hebra de ADN salir de la Tierra y llegar a la Luna varias veces. ¿Cómo comprender entonces que esa cadena no tuviera inscrita una enorme cantidad de genes? Y la siguiente pregunta, todavía más enigmática: si ahí no teníamos tantos genes… ¿qué estaba alargando tanto la cadena, y ocupando tanto espacio?
Una breve historia del ADN a la genómica
1871
Friedrich Miescher publica un artículo en el cual identifica la presencia de nucleína (ahora conocida como ADN) y proteínas asociadas en el núcleo de la célula.
1910
Albrecht Kossel gana el primer Premio Nobel en medicina por descubrir las 5 bases nitrogenadas: adenina, citosina, guanina, timina y uracilo.
1950
Erwin Chargaff descubre en muestras de ADN que la base adenina siempre se empata con la timina y que la citosina siempre se empata con la guanina.
1953
James Watson y Francis Crick, con contribuciones de Rosalind Franklin y Maurice Wilkins, descubren la estructura de la doble hélice del ADN.
1961
Marshall Nirenberg y Har Gobind Khorana identifican que las bases en el ADN se leen en bloques o codones: cada codón especifica un aminoácido que se agrega a la proteína en la síntesis.
1972
Susumo Ohno acuña el término “ADN basura”.
1980
Fred Sanger, Walter Gilbert y Paul Berg comparten el Premio Nobel de Química por desarrollar métodos de secuenciación del ADN.
1990
Se lanza el Proyecto del Genoma Humano, que busca secuenciar 3 000 millones de bases del genoma humano en 15 años.
1993
Phillip Allen Sharp y Richard Roberts ganan el Premio Nobel por descubrir que los genes del ADN se conforman de intrones y exones.
1995
Se secuencia el primer genoma de una bacteria, Haemophilus influenza.
1996
Se publica el genoma de la levadura, Saccharomyces cerevisiae. Nace Dolly, el primer animal clonado.
2002
Se lanza el Proyecto Internacional HapMap Project para producir un catálogo de variaciones genéticas humanas comunes y su ubicación en el genoma.
2003
Con 2 años de anticipación se completa la secuenciación del genoma humano.
Nace el proyecto ENCODE con el fin de identificar y caracterizar todos los genes en el genoma humano.
2012
ENCODE publica 30 artículos describiendo las regiones activas del genoma humano incluyendo la confirmación de que cuenta con 20 687 genes codificadores de proteínas.
2013
La Corte Suprema de Estados Unidos decreta que el ADN natural no se puede patentar.
2016
Se secuencia el primer genoma en el espacio a bordo de la Estación Espacial Internacional.
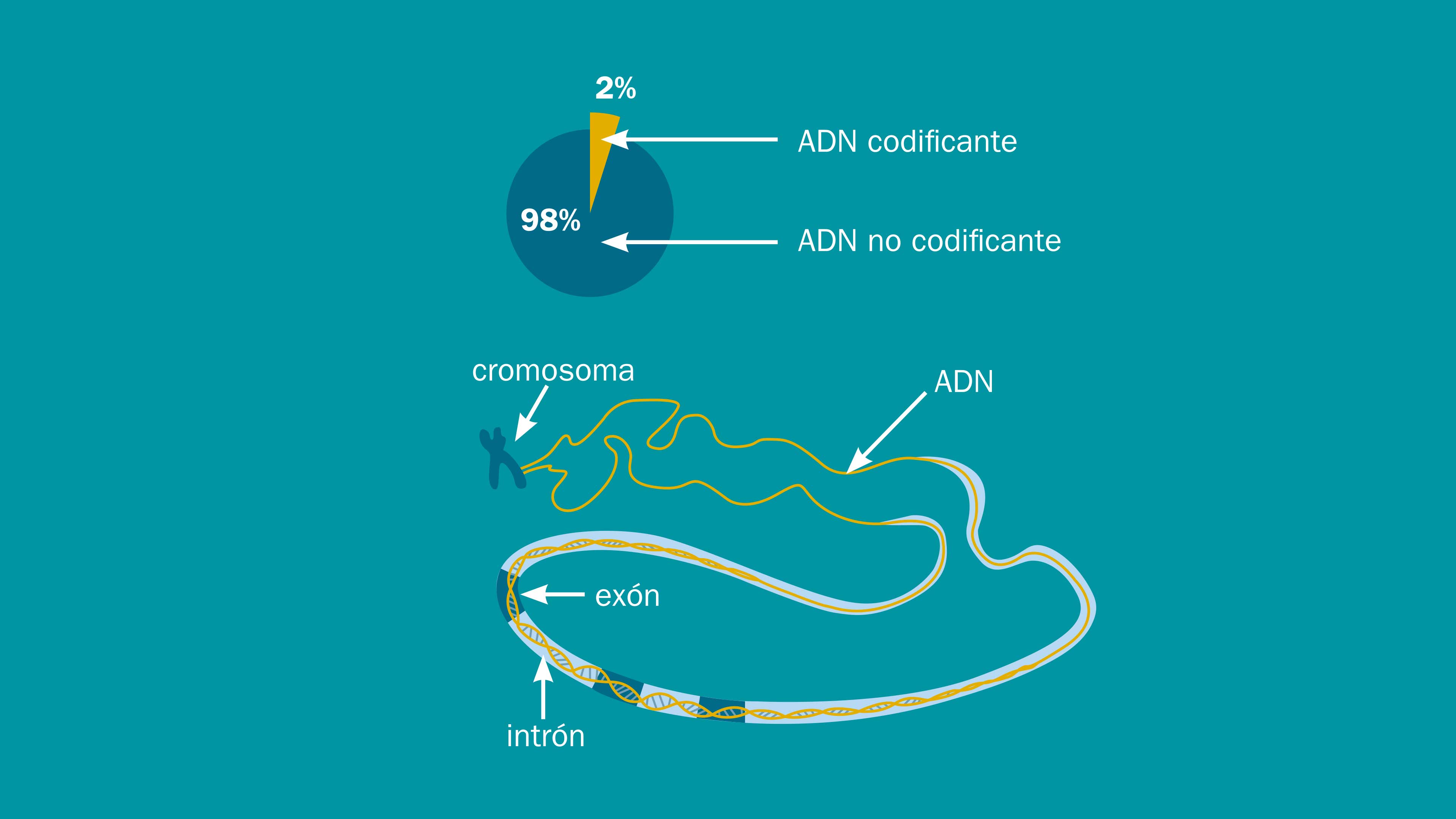
Ponga la basura en su lugar
Hoy sabemos que los genomas de los seres vivos vienen en tamaños que no están relacionados con la complejidad de estos últimos. En otras palabras, lo que vemos a nivel del individuo no corresponde necesariamente con el material genético que existe en el ambiente celular.
Las investigaciones sobre el genoma humano mostraron que apenas entre el 1 y el 2% de nuestro material genético está compuesto por segmentos de ADN que tienen genes con los códigos necesarios para fabricar proteínas. Estos segmentos reciben el nombre de exones o ADN codificante. El resto, que suma un inquietante 98% del material genético, corresponde a los intrones, cuya definición más simplista es que son los segmentos de ADN que no codifican para generar alguna proteína. En realidad, los intrones son mucho más versátiles, y no han sido tan sencillos de clasificar.
Al suponer que el ADN “útil” era solamente aquel con instrucciones para fabricar proteínas, los investigadores se enfrentaban al restante 98%, que resultaba un enigma evolutivo. A principios de los años 70, el biólogo evolucionista Susumu Ohno popularizó el término “ADN basura” para referirse a estos enormes segmentos de adeninas, timinas, citosinas y guaninas que al parecer, no servían para nada.
Inicialmente, Ohno inventó el término para designar a los pseudogenes, que son productos fallidos de la duplicación genética. Ya sea por mutaciones o por errores en el ensamblaje en la secuencia de nucleótidos, durante su duplicación el gen en cuestión queda incapacitado para codificar su proteína correspondiente, dando lugar así a una secuencia que se parece mucho al gen original, pero que no cumple ya con las funciones esperadas. El número de estos “accidentes” evolutivos no es menor: la lectura del genoma humano arrojó cerca de 20 000 de estos “esqueletos”, que siguen copiándose y aparecen en las cadenas de ADN de las generaciones siguientes. Para Ohno, esta dinámica de duplicar la “basura” carecía de sentido, y parecía una pérdida de energía evolutiva. Quienes opinaban como él, sugerían que estos segmentos no codificantes son un residuo evolutivo, un “estorbo bioquímico” que se ha quedado inmerso entre los sectores que sí son relevantes para la biología de la especie. El mismo Francis Crick se expresó a favor de esta visión en 1980, cuando publicó en la revista Nature un artículo con el sugerente título “El ADN egoísta: el máximo parásito”. Por su parte, en el libro Genoma: la autobiografía de una especie en 23 capítulos, el divulgador de la ciencia Matt Ridley comparó el genoma humano con un libro que se escribe en 23 capítulos, que corresponden con los 23 pares de cromosomas que tenemos como especie. Ridley nos invita a imaginar que en cada capítulo se cuentan historias diferentes, es decir: en cada cromosoma se ubican genes determinados que manifiestan a nivel del organismo funciones distintas. Para Ridley, los segmentos no codificadores del ADN son como los anuncios en las revistas: puede que tengan alguna función, pero en conjunto interrumpen la lectura. Esto es, en cierto modo, lo que sucede dentro de las células eucariontes, como las nuestras. El tamaño de la cadena de material genético tiene muchos segmentos que interrumpen la lectura entre los genes. En cambio, las células procariontes tienen una lectura más fluida; aunque son organismos unicelulares en apariencia muy simples, la lectura de su ADN resulta mucho más eficiente, pues su material genético “flota” en forma de anillo en el ambiente celular, y en éste se encuentran todos los genes de manera prácticamente continua. Utilizando la analogía de Ridley, en esas células hay poco espacio pero también pocos anuncios, por lo que la información se optimiza.
Genes huérfanos
Si bien el término “ADN basura” resultaba atractivo y de fácil adopción por el público, muchos investigadores se opusieron firmemente a su uso, argumentando entre otras cosas que la evolución biológica tiende a ser bastante económica y mantener segmentos enormes de ADN sin sentido no es típico del proceso evolutivo de las especies. Estos investigadores sugerían que el ADN aún guarda secretos y que la llamada “basura” en realidad se correspondía con segmentos cuya función no comprendimos a cabalidad en el inicio del estudio genómico. En esos segmentos de material genético se reporta la existencia de información vital para que los genes se expresen correctamente, y a tiempo. Al dejar de considerar al ADN que codifica proteínas como el único segmento interesante, los investigadores obtienen una comprensión integral del material genético en todos los seres vivos. La evidencia indica que en el mal llamado “ADN basura” se marca el ritmo de la expresión genética, mediante silenciadores y promotores que dan la pauta a los genes para actuar o dejar de hacerlo.
La longitud de nuestra cadena de ADN, compuesta por 3 000 millones de nucleótidos, da un filamento largo y dentro de 1 sola célula podemos guardar 2 m de este tesoro biológico. Si sumamos esos 2 m de ADN por cada célula del cuerpo humano alcanzamos una longitud de cadena apabullante, que permitiría a la hebra de ADN salir de la Tierra y llegar a la Luna varias veces.
Un descubrimiento reciente que proviene del “basurero” genómico, se refiere al nacimiento de los genes. Estos, como las personas, tienen familias con las que comparten ciertas afinidades y semejanzas, tanto en función como en composición bioquímica. Al igual que en un árbol genealógico, para cada especie se pueden rastrear sus ancestros bioquímicos. Cuando se estudia el proceso de la aparición de un gen “en reversa” se puede llegar, en principio, al gen original del que se desprenden éste y otros genes parecidos. Dado que las familias de genes con instrucciones para codificar proteínas útiles para la vida están presentes en especies biológicamente muy diferentes, no resultaba aventurado suponer que todo provenía de un conjunto de genes iniciales. Estos bloques de construcción primigenios comparten, por lo tanto, ancestros para todos los seres vivos y sus orígenes se remontan a aproximadamente 3 000 millones de años. En teoría, a partir de estos genes iniciales y sus combinaciones, mutaciones o cambios aleatorios se obtendría la diversidad de genes que podemos ver en las especies en la actualidad.
Hacia 1990, los científicos se enfrentaron con un enigma biológico: al menos la tercera parte de los genes en las levaduras tenía un origen desconocido, pues no había manera de relacionarlos con ninguna de las familias genéticas conocidas para la vida en el planeta. ¿De dónde provenían estos genes huérfanos? Inicialmente, se consideró que todavía nos faltaban muchas familias por encontrar por lo que cuando se tuviera el panorama completo, los genes huérfanos entrarían fácilmente en alguna categoría. Sin embargo, estas familias no fueron descubiertas al completar la lectura de los genomas de la levadura y al poco tiempo se reportó algo similar para el genoma de la mosca de la fruta, el ratón, los gorilas y los seres humanos. Hay genes nuevos, sin aparente relación con las familias de genes primordiales.
Para el Dr. Erich Bornberg-Bauer, de la Universidad de Münster en Alemania, si las partes nuevas del genoma sólo podían provenir de las partes viejas, no se podía explicar la diversidad biológica que encontramos en las especies. En su opinión, lo nuevo tenía que originarse de áreas diferentes del genoma, que no contuvieran los genes funcionales conocidos.
En 2006, David Begun de la Universidad de California propuso en un artículo publicado en la revista Genetics que ciertos genes de diferentes especies de mosca de la fruta “nacían” de secuencias de ADN no codificantes, que podían mutar para transformarse en genes funcionales. Su artículo generó polémica rápidamente. Quienes lo criticaban sostenían, por ejemplo, que para afirmar que un gen es nuevo, se tienen que conocer todos los genes viejos. Pero en el 2009, los resultados obtenidos por Diethard Tautz del Instituto Max Planck de Biología Evolutiva le dieron la razón a Begun. Tautz era originalmente un opositor de la hipótesis de Begun, pero al rastrear el origen para el gen Pldi en ratones, encontró que la región de la que provenía era un segmento de ADN “basura”, y que una serie de mutaciones habían transformado el segmento en un gen funcional que actualmente estaba involucrado en el tamaño de los testículos de los machos de la especie. Éste, y otros resultados semejantes, están generando una atención inusitada en estas regiones del genoma que antes se consideraban simple relleno.
En el 2015, la Dra. Mar Alba presentó hallazgos muy interesantes ante la Sociedad de Biología Molecular y Evolución de Viena. Alba y su equipo sorprendieron a la comunidad al reportar la existencia de 600 genes humanos que, se presume, emergieron del ADN no codificante. Generalmente, los genes nuevos son pequeños y codifican proteínas con tamaños moleculares modestos, por lo que pueden pasar desapercibidos; poco se sabe de su función, por lo que su estudio nos promete muchas sorpresas para el futuro.
La evidencia indica que en el mal llamado “ADN basura” se marca el ritmo de la expresión genética, mediante silenciadores y promotores que dan la pauta a los genes para actuar o dejar de hacerlo.
Una joya en la basura
Conforme avanza el siglo XXI, el “ADN basura” cobra sentido e importancia. El Proyecto ENCODE (siglas en inglés de “Enciclopedia de elementos del ADN”) es uno de los bastiones para valorar los segmentos de ADN que si bien no codifican proteínas, tienen otras funciones que empezamos a detectar. A partir del 2007, el proyecto superó la fase piloto e inició investigaciones para identificar y clasificar todos los elementos funcionales del ADN, extendiendo esta definición más allá de los genes que producen proteínas. ENCODE ha registrado las regiones de ADN que se muestran activas para la transcripción y regulación de la información genómica, y se ha dado a la tarea de localizar las zonas que producen ácido ribonucleico (ARN). El Dr. Ewan Birney, quien encabezó al grupo de 440 científicos en 32 laboratorios a nivel mundial para ENCODE, considera que el término “ADN basura” debe desecharse pues nos da una idea equivocada de la riqueza del genoma y su funcionamiento. A partir de los resultados obtenidos por ENCODE, Birney enfatiza que debemos mirar a otras regiones del ADN para responder preguntas sobre evolución o medicina genómica. En su opinión, “los segmentos con genes que codifican proteínas abarcan apenas un 2%, mientras que los segmentos que regulan la expresión de estos genes en el supuesto ADN basura pueden abarcar desde el 10 hasta el 80% del ADN restante”. Sería lógico aventurar, por lo tanto, que muchas respuestas vendrán de dichos segmentos, aunque ese rango es tan amplio que una parte importante del ADN podría resultar, de todas formas, inservible.
Al margen de las discusiones acerca del porcentaje funcional que tiene el ADN del ser humano, las recientes investigaciones de James Noonan de la Universidad de Yale, parecen darle la razón al grupo ENCODE. Noonan se dedicó a estudiar un segmento de “ADN basura” llamado HACNS1 en distintas especies de mamíferos. Al comparar este segmento del ADN del ser humano con el de otras especies, Noonan y su equipo detectaron que el HACNS1 humano ha acumulado un número importante de mutaciones a lo largo del tiempo y se ha modificado tanto con respecto al genoma de otros animales que, en palabras de Noonan, “se puede considerar característico y único de los humanos”. Si esta parte del ADN es simple basura, y no sirviera para nada, las mutaciones no generarían impacto alguno en el genoma. Para averiguarlo, los investigadores marcaron el HACNS1 humano con sustancias que muestran color cuando partes del mismo se activan y lo colocaron en embriones de ratón. Noonan y sus colegas encontraron que el HACNS1 no era el desecho que se creía: el color apareció en las partes que dan lugar a las patas de los ratones, específicamente, en las que están involucradas en el desarrollo de los pulgares. Sabemos que uno de los hitos evolutivos de nuestra especie se presentó cuando conseguimos ponernos de pie y comenzamos a manejar los pulgares para agarrar objetos. En los humanos el HACNS1 podría haber funcionado como uno de los directores de orquesta para que ciertos genes modificaran poco a poco la forma de los pies y de las manos, separándonos de otros primates.
A partir de éste y otros descubrimientos semejantes, el ADN basura comienza a contemplarse bajo otra luz. Una luz, por cierto, mucho más interesante. Además, la comprensión del genoma en su totalidad se vuelve trascendental pues de poco sirve conocer los 20 000 genes que nos conforman si no tenemos idea precisa de qué los incita a expresarse. Las últimas investigaciones indican, por lo tanto, que llegó el día de reivindicar a la basura.
Más información
- Beyer Ruiz, Maria Emilia, Gen o no gen: el dilema del conocimiento genético, Lectorum, México, 2002.
- Díaz, Alberto y Golombek, Diego; ADN: cincuenta años no es nada, Siglo XXI, Buenos Aires, 2007
- Centro de Ciencias Genómicas: www.divulgacion.ccg.unam.mx
María Emilia Beyer es divulgadora de la ciencia y bióloga de profesión. Es pasante de la maestría en Filosofía de la Ciencia de la UNAM. Actualmente trabaja en la Dirección General de Divulgación de la Ciencia de la UNAM








