


Los coronavirus mutantes: ¿me debo preocupar?
Miguel Ángel Cevallos
Ilustración: Wetzkaz Graphics/Shutterstock
Cómo surgen y qué efectos pueden tener las nuevas variantes del SARS-CoV-2, causante de la pandemia que hoy vivimos, en la propagación de los contagios, la condición de los pacientes y la eficacia de las vacunas que se están aplicando.
¡Renovarse o morir! Ningún dicho mejor que este para comprender la biología de los virus. Lo único que hacen estos microscópicos patógenos es multiplicarse. Para lograrlo necesitan un huésped al cual infectar, evadir su respuesta inmune y replicarse dentro de sus células lo más eficientemente posible. En el proceso de replicación surgen variantes del virus a través de mutaciones en su material genético. Las variantes que van a prevalecer son aquellas que se repliquen a mayor tasa y que eludan mejor el sistema inmunitario del huésped. Los coronavirus, al ser virus cuyo genoma es una molécula de ARN, mutan más frecuentemente que sus contrapartes virales con un genoma de ADN. Esto, aunado a las enormes progenies que se producen en cada ciclo de infección, hace que los coronavirus evolucionen rápidamente; se ha calculado que adquieren una nueva mutación cada 11 días. Las mutaciones —errores que se producen cuando se copia el material genético para la replicación del virus—, pueden aparecer en cualquier región del genoma y su destino depende de los cambios que inducen en la partícula viral que los porta. Si un coronavirus adquiere una mutación que “estropea” un gen con las instrucciones para que la célula infectada fabrique alguna de las proteínas de la superficie del virus, o que ponga en jaque su habilidad para replicarse, seguramente se extinguirá con rapidez. En contraste, aquella partícula viral que adquiera una mutación que le permita reconocer a la célula huésped mejor que sus congéneres y/o que pueda evadir mejor nuestro sistema inmunitario, dejará más descendencia y, por esta razón, será la variante que predominará en algún momento. También pueden producirse mutaciones que ni fu ni fa; es decir, que no le confieren ninguna ventaja al virus, pero tampoco una desventaja. Esto se debe a que algunas mutaciones pueden modificar un gen sin alterar la estructura de la proteína que se produce o su función. A estas mutaciones se les califica como neutrales.
Vigilancia cuidadosa
Poder determinar la secuencia de los genomas virales de una manera rápida y barata es fundamental para analizar, en tiempo real, la dinámica de la pandemia que estamos sufriendo. La primera secuencia del genoma del SARS-CoV-2 se obtuvo pocos días después del inicio de la pandemia de COVID-19 y fue la base para dar inicio al diseño de las vacunas que hoy se están aplicando. Pero esto no acaba ahí; todos los días se obtienen secuencias nuevas. Hasta ahora se puede consultar en bases de datos internacionales la secuencia de más de 50 000 genomas completos del virus y cientos de miles de secuencias parciales (de cachitos del genoma del virus). A pesar de que hay muchísimas secuencias depositadas en esas bases de datos, la contribución de los países a esta tarea es bastante desigual. La mayor parte de las secuencias provienen del Reino Unido, Estados Unidos, Islandia, Australia y Holanda. En comparación, México, a través del InDRE (Instituto de Diagnóstico y Referencia Epidemiológicos), ha hecho su esfuerzo depositando poco más de 800 genomas, al menos en la fecha en que estoy escribiendo este artículo.
Quizá la base de datos de libre acceso más grande y actualizada del SARS-CoV-2 es la que sostiene la iniciativa GISAID (www.gisaid.org). Esta iniciativa, que funciona con recursos públicos y privados, se creó originalmente en 2008 con el fin de monitorear el virus de la influenza. Su sitio de internet cuenta no solo con las secuencias totales y parciales de los coronavirus aislados en todo el mundo durante el transcurso de la pandemia, también con las herramientas bioinformáticas adecuadas para analizar dónde surgen las nuevas variantes, qué tan bien se trasmiten, cómo “migran” de una ciudad a otra, e incluso determinar dónde y cuándo se extingue una nueva variante y cuál la sustituye.
Para identificar las diferencias entre las variantes del SARS-CoV-2 que circulan actualmente, se usa como referencia el genoma del virus de Wuhan.
Con el análisis de los genomas obtenemos una visión sobre la dinámica de la epidemia y además información valiosísima sobre la biología básica del virus, como la que nos permite identificar qué cambios hacen que una variante sea más eficiente que otra. También podemos rastrear de mejor manera las cadenas de infección y hacer una vigilancia epidemiológica más certera. Para que quede más claro cómo se usan estos datos, mencionaré un ejemplo que publicó un grupo de investigadores encabezados por el Dr. Barnaby E. Young, en la revista The Lancet en agosto de 2020. En ese artículo se describe una variante del virus, llamada D382, que surgió en Wuhan, China, y a la que le hacía falta un pedacito de ARN; esta variante carece de uno de los genes del virus original (ORF7b) y el que le sigue (ORF8) no funciona. La D382 se propagó eficazmente por un tiempo, e incluso llegó a Singapur. De hecho, en ese país, en enero y febrero de 2020, se detectó a un grupo de pacientes infectados con esa variante, pero la enfermedad que producía era más bien leve; los enfermos usualmente no requerían oxígeno suplementario y presentaban una reacción inflamatoria moderada. Se sabe que esta variante ha surgido, de manera independiente, en otras regiones del mundo causando en los pacientes los mismos efectos. Con ello, podemos vincular los genes faltantes con la capacidad del virus de escapar a la respuesta inmunitaria del huésped. Con un adecuado diagnóstico molecular (en el que se detecta ARN del virus en células del paciente) se podría predecir que los infectados con virus que portan esta mutación tienen un buen pronóstico y podrían sin problemas sobrellevar la enfermedad en casa.
Luces en el horizonte
Muchos de los esfuerzos que han hecho los gobiernos y las farmacéuticas para combatir la pandemia se han centrado en generar vacunas, lo más pronto posible. Sin embargo, las investigaciones sobre fármacos que pudieran contender con la infección del coronavirus se han quedado atrás y estamos perdiendo armamento importante para lidiar con el virus. Recordemos que, en el 2009, con la pandemia de influenza AH1N1 contamos con un antiviral muy eficiente, el Tamiflú, que disminuyó notablemente los estragos que pudo haber causado esa pandemia. Afortunadamente, hay nuevas luces en el horizonte. En enero de este año se publicó en la revista Science una investigación hecha por un consorcio multinacional, encabezado por el Dr. Adolfo García Sastre, del Hospital Monte Sinaí, en Nueva York, que muestra en ensayos preclínicos, que el fármaco Plitidepsina, fabricado por la compañía española PharmaMar, es sumamente eficaz inhibiendo la replicación del virus. De hecho, es 100 veces más potente que el Remdesivir, que es el antiviral que actualmente se usa en los hospitales para ayudar a combatir las infecciones de SARS-CoV-2. La Plitidepsina es un medicamento antitumoral que actúa sobre una proteína humana llamada eEF1A que es muy importante no solo en la síntesis de las proteínas, también ayuda al buen funcionamiento de nuestras células. Una de las cosas que hace muy interesante a la Plitidepsina es que actúa sobre una proteína humana y no una viral, lo cual promete que será también muy útil en inhibir la replicación de los nuevos linajes de coronavirus. No hay que cantar victoria, estas primeras observaciones se hicieron en ensayos preclínicos, usando ratones como modelo experimental. Al igual que las vacunas, los fármacos también tienen que pasar las Fases I, II y III de los ensayos clínicos para que se apruebe su uso.
Súper linaje
Los expertos en epidemiología del SARS-CoV-2 suelen agrupar las variantes que van surgiendo durante el transcurso de la epidemia en linajes y el modo en que lo hacen recuerda la manera en que se construyen las genealogías familiares: en la medida en que dos individuos comparten un mayor número de mutaciones, más emparentados estarán. Se considera que dos variantes virales pertenecen a un mismo linaje si comparten un conjunto de mutaciones que heredaron de sus antecesores. Para identificar las diferencias entre los virus, los epidemiólogos siempre usan como referencia el genoma del primer virus que se secuenció en los primeros días de la epidemia, en Wuhan, y del cual se originaron todas las variantes que circulan el día de hoy.
Los epidemiólogos ingleses observaron en diciembre del año pasado, en la ciudad de Kent, un incremento muy pronunciado en el número de infecciones, a pesar de las estrictas medidas de cuarentena que se estaban tomando. El análisis de los genomas de algunas muestras de ese particular repunte epidémico determinó que los virus responsables pertenecían a un nuevo linaje, que llamaron B.1.1.7. El estudio también reveló que este virus surgió en septiembre de ese mismo año y que en un inicio circuló a bajos niveles, pero un par de meses más tarde era la variante preponderante en esa región y poco después desplazó a otras variantes del país. Ahora se ha detectado su presencia en al menos 50 países y seguramente se convertirá en la variante dominante en todo el orbe. Los miembros de este linaje comparten 23 mutaciones. Pero quizá lo más relevante es que muchos de estos cambios ocurren en el gen con las instrucciones para fabricar la espícula, la proteína crucial para que comience el proceso de infección del virus.
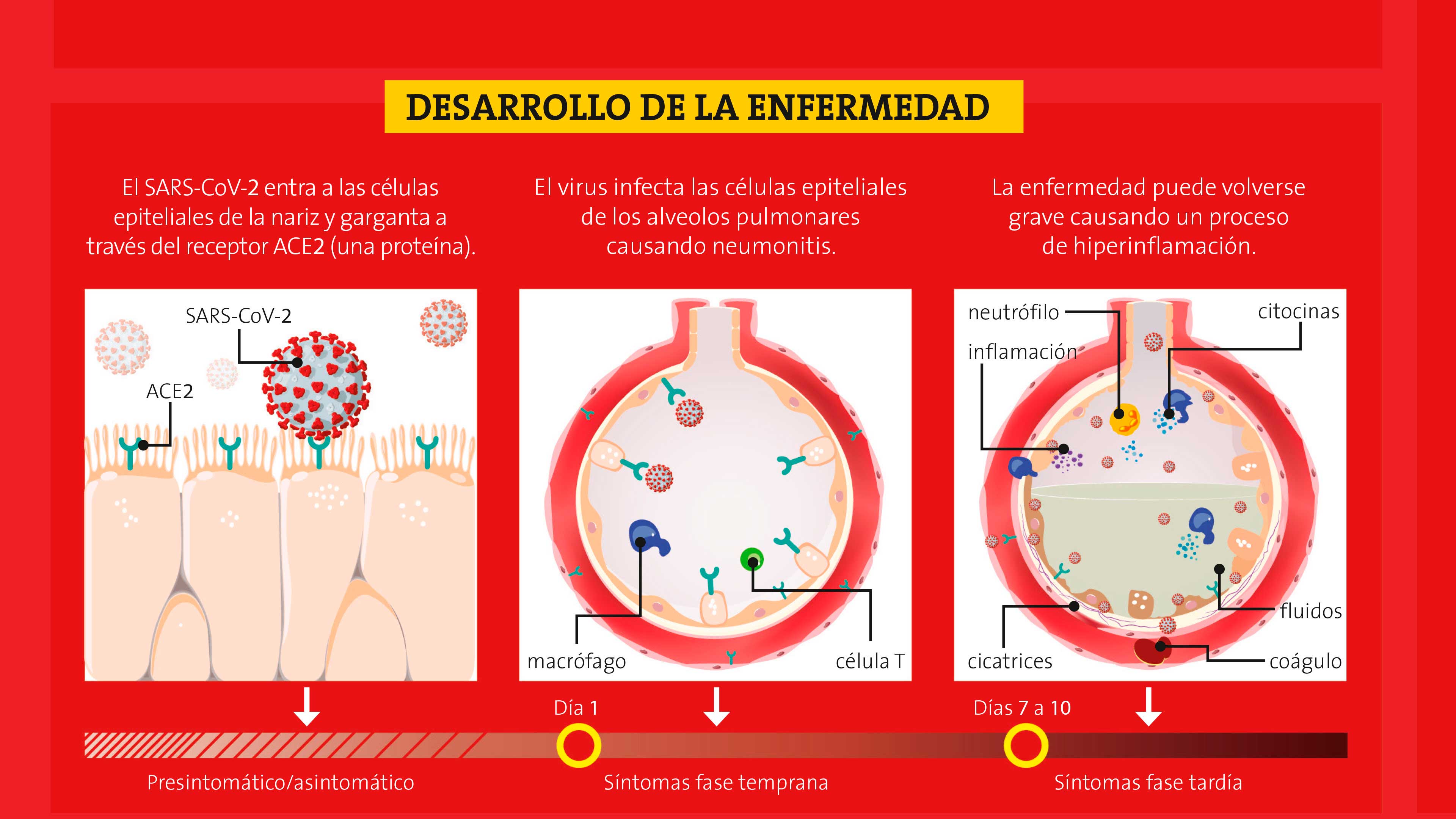
El diablo se esconde en los detalles
La espícula es una de las proteínas de la superficie del virus y la responsable de reconocer la proteína ACE2, el receptor del virus, que se encuentra en la superficie de las células epiteliales que recubren nuestro aparato respiratorio. La interacción que ocurre entre la espícula y la ACE2 es el evento que da inicio el proceso de infección. En una analogía que se trae a colación frecuentemente, se concibe a la espícula como una llave y a la ACE2 como la cerradura de una puerta que se abre al interior de la célula. Así es que cualquier cambio en la estructura de la espícula podría tener un fuerte impacto en el proceso de infección, por esta razón los epidemiólogos están atentos a cualquier alteración que ocurra en el gen con las instrucciones para producirla (en un lenguaje más técnico, el gen que la codifica).
El gen de la espícula en el linaje B.1.1.7 porta ocho mutaciones. Dos de ellas son pequeñas deleciones (le faltan dos pedacitos) y las otras seis cambian la secuencia de la proteína. Entre ellas, tres son las más preocupantes: la primera se conoce como N501Y y altera una región esencial de la espícula, que en la analogía corresponde a los dientes de la llave. Una manera de verlo es pensar que la mutación convierte la llave en una ganzúa que abre con más facilidad la cerradura ACE2. De hecho, se ha demostrado experimentalmente que esta mutación hace que la interacción espícula-ACE2 sea mucho más fuerte. Adicionalmente, una de las deleciones que tiene este gen elimina un pequeñísimo segmento de la espícula (dos aminoácidos), que permite a los virus que lo portan evadir más fácilmente nuestro sistema inmunitario, fortaleciendo así su capacidad de contagiar. La tercera mutación de interés modifica la vecindad inmediata de una región de la espícula que se conoce como sitio de corte de la Furina, que es muy importante en el proceso de infección. La mutación en este sitio podría potenciar la capacidad de infección del linaje B.1.1.7. Pero aún falta hacer experimentos en el laboratorio para comprobarlo.
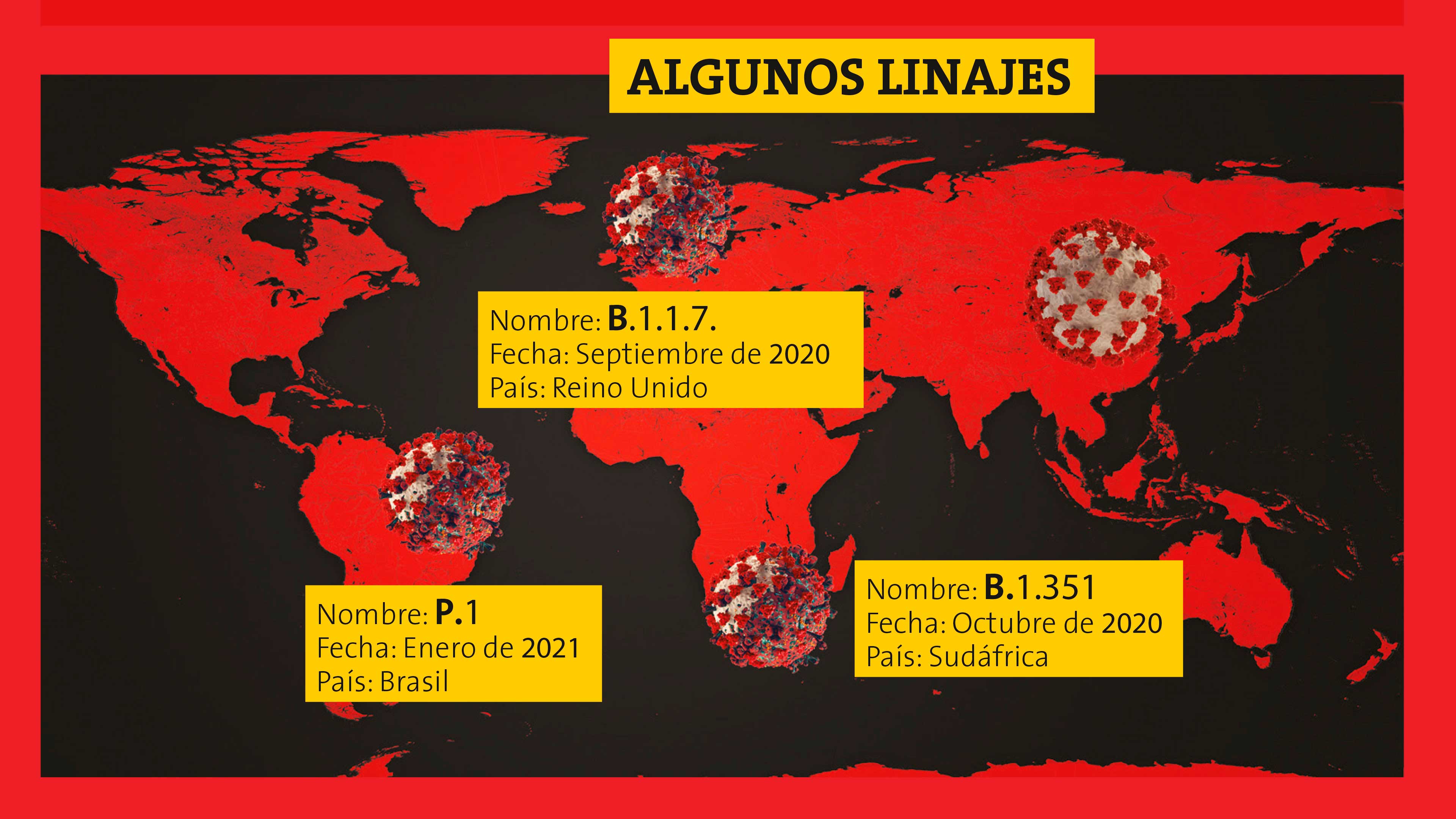
Otros linajes que preocupan
Hay otros linajes recientes que se consideran potencialmente peligrosos. Uno de ellos, el llamado B.1.351, apareció en Sudáfrica en octubre del año pasado y es el responsable de un notable incremento en los casos de COVID-19 en ese país. Este linaje también porta cambios en la espícula incluyendo la mutación N501Y ya mencionada y otra mutación llamada E484K que según informes recientes otorga al virus cierta resistencia a las vacunas. Otro linaje, el P1, surgió probablemente en Manaos, Brasil, y es el causante de un alto porcentaje de las infecciones en esa ciudad, probablemente la más golpeada del planeta. Este linaje posee tres mutaciones en su espícula: la N501Y y la E484K, y la llamada mutación K417T, cuyo impacto en la biología del virus es todavía desconocido. Hay otra variedad que surgió en España y que solo porta una mutación en la espícula (A222), pero hace que nuestros anticuerpos no bloqueen con agilidad la infección.
¿Cómo se nombran las mutaciones?
Para saber si ha ocurrido una mutación en una muestra de SARS-CoV-2, hay que secuenciar el genoma de la muestra de interés y compararla con la secuencia de referencia (Wuhan-Hu-1) que se obtuvo de un virus los primeros días de la epidemia. Hay dos maneras de darle nombre a las mutaciones: la primera es basarse en la posición del genoma en que ocurrió la mutación y la otra es nombrarla refiriéndose al cambio que pudiera inducir en la proteína afectada. Empecemos por la primera: si el genoma de referencia tiene un tamaño de 29 903 nucleótidos y la mutación sucedió en la posición 24380 y cambió un nucleótido citosina (C) por una adenina (A), la forma correcta de nombrarla sería C24380A. La otra manera de nombrar una mutación es a partir de la proteína que afectan. Las proteínas están compuestas por 20 diferentes aminoácidos y cada aminoácido tiene una abreviación de una sola letra. Si una mutación afecta el gen que codifica la espícula (S) y cambia el aminoácido asparagina (N) por una valina (V) en la posición 501 de los aminoácidos que conforman esta proteína, se denomina N501V.
¿Me debo preocupar por las nuevas variantes?
La respuesta a esta pregunta es sí y no. Pero empecemos por el sí: se estima que estos nuevos linajes son mucho más eficientes infectando que el resto. En particular, se ha encontrado que el linaje B.1.1.7 es un 50 % más transmisible que el virus original. Es muy probable que estas variantes, en cuanto se importen a una región determinada del planeta, desplazarán rápidamente a todos los linajes que estén circulando ahí. En otras palabras, veremos un pico en el número de casos y de muertes en todas aquellas regiones en las que estos linajes se hagan presentes.
Otro aspecto a considerar es que hay poquísimos fármacos desarrollados para combatir el SARS-CoV-2. Entre ellos están los dos anticuerpos monoclonales de la compañía Lilly diseñados específicamente para bloquear la espícula del virus, de manera que cualquier cambio en la estructura de esta proteína podría reducir su eficacia.
Por todo lo anterior, es más que evidente que debemos redoblar las precauciones para evitar infectarnos y esto implica seguir usando cubre bocas y, si se puede, acompañado de una careta. No debemos cejar en la llamada sana distancia; procurar una buena ventilación en espacios cerrados; lavarnos las manos frecuentemente o usar gel desinfectante con alcohol al 70 % y aislarnos por dos semanas si mostramos signos de una infección respiratoria.
Sigamos con el no me debo preocupar (demasiado): los pocos datos con que contamos, quizá demasiado prematuros, todavía sugieren que las infecciones que inducen los nuevos linajes no son ni más ni menos agresivas que las que actualmente padecen los enfermos de COVID-19. En otras palabras, estos linajes son más infecciosos que el resto de los linajes de SARS-CoV-2 en circulación, pero, al parecer, igual de virulentos.
La mayor parte de las vacunas que se han desarrollado se basan directa o indirectamente en bloquear la espícula. Cualquier mutación que altere la estructura de esta proteína es de especial preocupación, como ya mencioné. Por esta razón, en el momento en que surgieron estos nuevos linajes, muchos investigadores se dieron a la tarea de determinar si las vacunas aprobadas o a punto de aprobarse, nos pueden proteger de los nuevos intrusos. Los científicos de la compañía Pfizer, encabezados por el Dr. Philip Dormitzer, junto con investigadores de la Universidad de Texas, descubrieron que las personas inoculadas con la vacuna de Pfizer generan anticuerpos que siguen neutralizando tanto a los virus que portan la mutación clave N501Y, como a los que no la portan: esa vacuna sigue siendo eficaz. La vacuna de la compañía Astra-Zeneca, la tercera aprobada para su uso, es muy eficiente con los linajes más antiguos, pero no tanto con los nuevos. De hecho, no sirve para controlar la variante que surgió en Sudáfrica y por esta razón se dejó de aplicar en ese país. Otras vacunas que ya concluyeron su fase III de estudio o están a punto de hacerlo, como la de la compañía Novavax, la de Johnson & Johnson y la rusa Sputnik V, al parecer muestran una tendencia similar, es decir funcionan muy bien con las viejas variantes y razonablemente bien con las nuevas. Con este nuevo escenario, las farmacéuticas están empezando a diseñar refuerzos de la vacuna para combatir mejor a las nuevas variantes.
La aplicación masiva de las vacunas, lo más pronto posible, mitigará los efectos de los nuevos linajes. Dadas estas circunstancias, probablemente nos vamos a enfrentar a evaluar los riesgos de empezar a usar vacunas que no han concluido la fase III. Es quizá necesario que un panel de expertos evalúe los datos de esa fase antes de que se publiquen. Pero esperar muchos meses, al ritmo en que están surgiendo los nuevos casos y la velocidad con que los nuevos linajes están sustituyendo a los viejos, costará literalmente miles de vidas.
Debemos redoblar las precauciones para evitar infectarnos, lo cual implica seguir usando cubre bocas.
Antes de concluir, me gustaría mencionar que la ciencia tiene caminos insospechados; en un artículo de la revista Science, de enero de este año, un grupo de investigadores del Instituto Tecnológico de Massachusetts, liderados por el Dr. Bryan Bryson, describe una nueva herramienta bioinformática que es capaz de reconocer, usando secuencias de genomas virales, cuáles son las variantes con el potencial de evadir el sistema inmunitario y volverse peligrosas, lo que agilizará la vigilancia epidemiológica de esta y otras pandemias Lo más interesante del artículo es que esta herramienta utiliza metodologías de disciplinas muy dispares que a primera vista no tienen que ver una con la otra, como la lingüística y la inteligencia artificial; esta última a través de los algoritmos del llamado aprendizaje automatizado (machine learning) que permite encontrar patrones en enormes cantidades de datos. Para terminar, quiero recalcar que para recibir la vacuna hay que estar vivo primero ¡cuídense!
- Khan Academy, “Evolución de los virus”: https://es.khanacademy.org Organización Mundial de la Salud,
- “Variantes del SARS-CoV-2”: www.who.int/csr/don/31-december-2020-sars-cov2-variants/es/
- Asociación Española de Vacunología, “El SARS-CoV-2 y sus mutaciones”: www.vacunas.org/el-sars-cov-2-y-sus-mutaciones/
Miguel Ángel Cevallos, frecuente colaborador de ¿Cómo ves?, es doctor en investigación biomédica básica y especialista en genética molecular bacteriana. Trabaja en el Centro de Ciencias Genómicas de la Universidad Nacional Autónoma de México.








