


Plegando proteínas
Alejandra Manjarrez

Ilustración: Shutterstock
La inteligencia artificial está revelando la estructura de moléculas esenciales para la vida
Durante la Segunda Guerra Mundial escasearon en los países en conflicto toda clase de productos, desde azúcar hasta jabón y zapatos. Uno de los alimentos que más se racionaron fue la carne. Ante la escasez de carnes tradicionales comenzó a investigarse la posibilidad de usar carne de ballena como suplemento (la guerra hace cosas muy raras). Gracias a esa investigación, a principios de la década de 1950 el bioquímico John Kendrew, de la Universidad de Cambridge, descubrió en su universidad una reserva congelada de carne de cachalote, una especie de ballena que se sumerge a profundidades enormes para buscar alimento.
Cachalotes y rayos X
Kendrew estaba interesado en determinar experimentalmente la estructura atómica de las proteínas. Hasta entonces nadie había logrado escudriñar la escala nanométrica que habitan las proteínas para apreciar su forma tridimensional. El problema era muy importante debido al papel esencial de estas moléculas —gigantescas para los estándares celulares— en el funcionamiento de un organismo. Conocer su forma con precisión proporciona una clave para entender cómo funcionan estas máquinas moleculares y cómo interactúan con otras moléculas, información útil para campos tan diversos como la neurobiología, la evolución y la inmunología. El investigador inglés había volcado casi todos sus esfuerzos en el estudio de una proteína en particular: la mioglobina, que almacena oxígeno en los músculos. Su intención era usar una técnica llamada cristalografía de rayos X, que aprovecha el fenómeno de la difracción de los rayos X cuando pasan a través de un cristal para revelar la estructura incógnita de la molécula. Para sus experimentos Kendrew necesitaba crear grandes cristales de mioglobina, lo que se puede hacer de varias formas, todas las cuales requieren la proteína en buenas cantidades.
Antes de su encuentro con la carne de cachalote refrigerada Kendrew había empleado mioglobina de corazón de caballos, pero pronto descubrió que los animales buceadores, que necesitan aguantar la respiración durante tiempos prolongados, tienen concentraciones más altas de esta proteína. Tras intentar con tortugas, delfines, pingüinos y otros animales marinos, el investigador consideró las posibilidades que ofrecían los cachalotes. En una carta a un colega Kendrew escribe: “hace tres semanas obtuvimos los cristales de mioglobina más maravillosos: de cachalote, ni más ni menos”. “Son cristales gigantescos”, añade, seguramente emocionado tras unos siete años de trabajar en este proyecto.
Con los cristales de mioglobina de carne de cachalote, Kendrew y su equipo lograron determinar que la proteína tenía una estructura formada por ocho hélices conectadas por bucles. Casi al mismo tiempo, su colega Max Perutz, en el mismo instituto, descifró la estructura tridimensional de una proteína hermana, la hemoglobina. A partir de las formas que revelaron estos equipos pudo entenderse, por ejemplo, cómo se une la hemoglobina a las moléculas de oxígeno en los pulmones, cómo las libera y cómo se las pasa a la mioglobina. En 1962 Kendrew y Perutz recibieron el premio Nobel de Química por sus descubrimientos, que les tomaron años, equipos completos de trabajo y miles y miles de libras. Más de seis décadas después, hay programas de inteligencia artificial que pueden hacer algo muy similar en cuestión de minutos, y hasta en segundos.
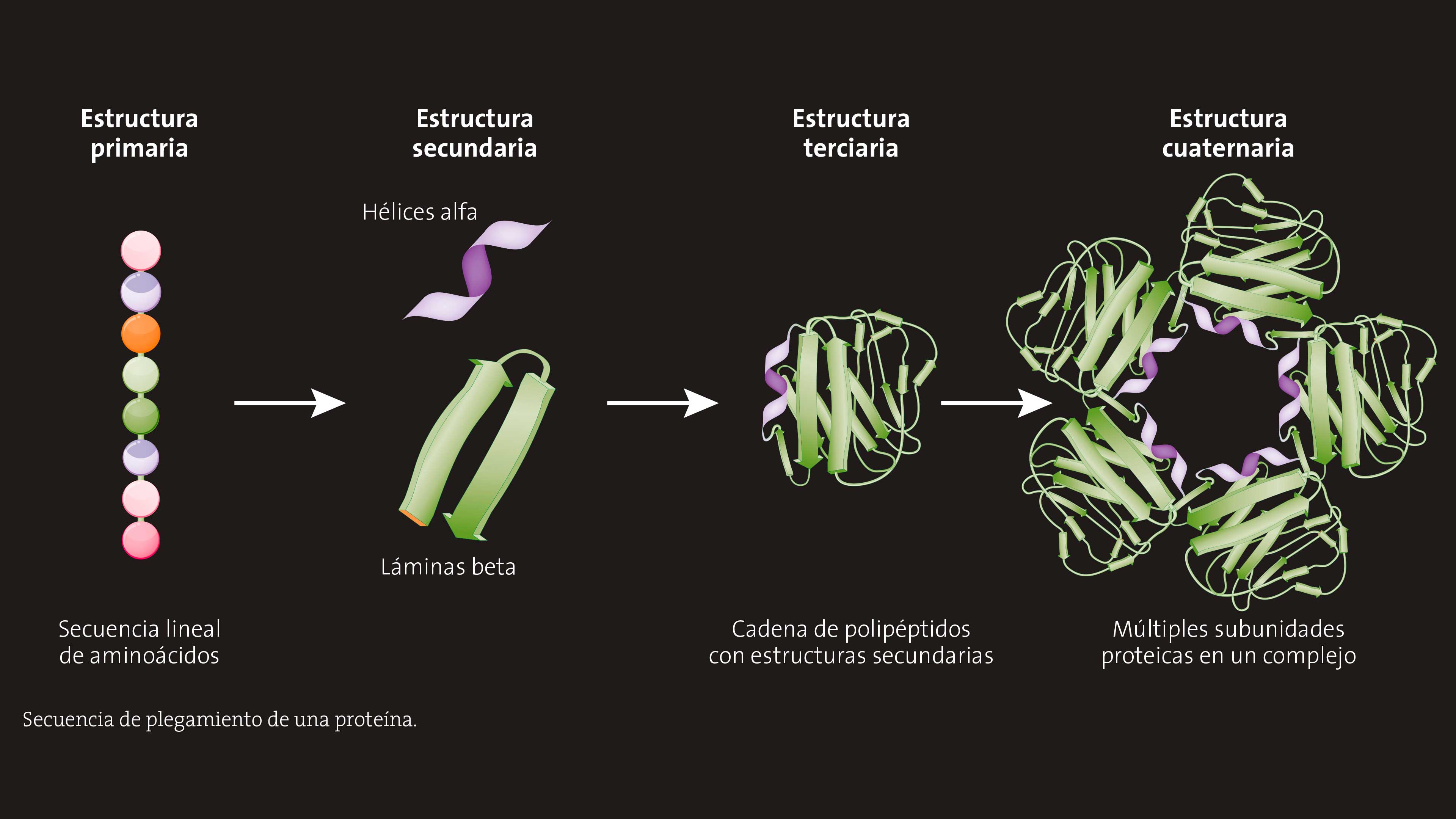
Un problema duro de roer
Desde aquellos trabajos pioneros se ha descifrado la estructura de montones de proteínas, y hemos descubierto que tienen una inmensa diversidad de formas. Unas parecen barriles o donas, otras bastones retorcidos y otras son más bien aplanadas. Las hay parecidas a una suerte de dardo —como la famosa espícula del virus sars-CoV-2—, las hay simétricas y las hay enmarañadas, pequeñas o pesadas.
Las proteínas están formadas por cadenas cuyos eslabones son moléculas llamadas aminoácidos. Existen 20 aminoácidos, de modo que la cantidad de combinaciones posibles es inmensa —lo mismo ocurre con las 27 letras del alfabeto, con las cuales puede crearse un abanico muy diverso de palabras (azahar, tintico, serendipia, febril, perenne, vals, esternocleidomastoideo)—. La forma de cada proteína está determinada por el orden específico de los aminoácidos, y esa cadena a su vez está codificada en el genoma de cada organismo. La secuencia de aminoácidos puede escribirse de forma lineal, pero para que la proteína funcione debe adoptar una forma tridimensional concreta; para esto la cadena se pliega de maneras muy complejas, que durante años fueron extremadamente difíciles de predecir a partir de la simple secuencia.
Una proteína promedio tiene entre 300 y 400 aminoácidos (aunque las hay mucho más grandes). Para entender cómo se va retorciendo y acomodando tridimensionalmente es necesario predecir cómo interactúan todos esos eslabones entre sí y con el medio acuoso que los rodea dentro de la célula. Por ejemplo, si entre los componentes de un aminoácido hay uno al que no le guste el agua, esa estructura se doblará hacia dentro de la proteína. También es necesario saber qué tipo de interacciones habrá entre los componentes de uno y otro y predecir si la interacción será débil o fuerte. Aunque hay mucha información que facilita ese trabajo, con tantos eslabones la labor sigue siendo muy compleja.
La tarea de descubrir los principios generales que rigen cómo se pliega una secuencia de aminoácidos determinada se conoce como el problema del plegamiento de proteínas, y desde hace décadas se sabe que es imposible resolverlo sin ayuda de computadoras. El biólogo germano-rumano Andrei Lupas, uno de los científicos que trabajan en este problema, lamentó varias veces que no viviría para ver su solución. Pero en una entrevista en noviembre de 2020 ya no parecía tan pesimista. Se acababa de anunciar que un programa desarrollado por la compañía británica de inteligencia artificial DeepMind de Google había arrasado en un desafío bienal en el que compiten diversos grupos de investigación para predecir la estructura tridimensional de proteínas a partir de su secuencia de aminoácidos. El programa se llama AlphaFold 2. Lupas fue juez de los modelos presentados ese año en la competencia, y aunque es cauto y no afirma que el problema se ha resuelto por completo, reconoció que este logro “lo cambiará todo”.
Predecir o posdecir
El concurso que ganó AlphaFold 2 en 2020 fue la edición número 24 del experimento casp (siglas en inglés de “Evaluación Crítica de la Predicción de Estructuras de Proteínas”).
Ya desde la década de 1990 había grupos de investigación que decían tener las herramientas y los modelos para predecir la estructura tridimensional de una proteína a partir de su secuencia de aminoácidos. Pero lo que hacían estos investigadores era comparar sus predicciones con estructuras que ya se conocían experimentalmente. En lugar de predicciones, Lupas las describe como “postdicciones”.
En 1994 un grupo de científicos dirigido por el biólogo computacional John Moult, de la Universidad de Maryland, organizó el experimento casp para poner a prueba estos modelos de manera más objetiva. Los aspirantes se enfrentaban al reto de descifrar proteínas cuyas estructuras ya estaban determinadas experimentalmente, pero que aún no habían sido publicadas. Es decir, los participantes debían trabajar a ciegas.
En aquel primer casp quedó claro que los modelos no funcionaban: ninguno alcanzó una precisión aceptable. Lupas añade que casi ninguno de los grupos que participaron en esa ocasión regresó a futuros casp. Tampoco hubo mucho éxito en el segundo encuentro, en 1996. En marzo del siguiente año, el periódico The New York Times lo resumió todo en este encabezado: “Diseñar vida: Proteínas 1, computadora 0”. El problema excedía las capacidades de las computadoras más potentes.
A partir de 1998, sin embargo, empezaron a notarse algunos avances. Se desarrolló una nueva manera de enfrentar el problema: buscarles a las proteínas de estructura desconocida “parientes” con una estructura conocida. A partir de la secuencia de aminoácidos de la proteína a descifrar se pueden identificar proteínas con secuencias parecidas. Y si se conoce la estructura tridimensional de éstas, puede usarse como modelo para inferir la que aún se desconoce. Esta técnica fue útil especialmente para predecir la estructura de proteínas de dificultad intermedia. Por ejemplo, en ese entonces Lupas estudiaba unas proteínas que ayudan a bacterias patógenas a adherirse a las células de los animales que infectan. Comparando las secuencias de una de estas familias de “adhesinas” con otras parecidas, el equipo pudo predecir su estructura, con ciertas limitaciones.
Con todo, el problema del plegamiento parecía aún estar lejos de resolverse.
Cambia el tablero con IA
En los experimentos casp que siguieron hubo algunos pequeños saltos, pero ninguno significativo, especialmente en las estructuras difíciles. Las calificaciones que se le otorgan a los modelos revelan un estancamiento todavía hasta 2016: en una escala de 100 puntos de precisión de la predicción, hasta ese año ningún equipo obtuvo una puntuación arriba de 40. En 2018, sin embargo, algo cambió. En el terreno de la predicción de moléculas difíciles las calificaciones subieron de 40 a más de 60. Muchos de los modelos computacionales recientes inauguraban una nueva estrategia para estudiar este problema: echar mano de redes neuronales artificiales, sistemas de cómputo inspirados en el cerebro humano y su cableado neuronal, muy útiles para identificar patrones en grandes cantidades de datos. AlphaFold, que ese año debutó con su primera versión en casp, fue el más exitoso.
Dos años después, en 2020, la segunda versión de AlphaFold arrasó en la competencia: la puntuación media de este algoritmo para la predicción de proteínas difíciles fue de 87 puntos. En unos pocos años la inteligencia artificial parecía haber cambiado el tablero en ese juego de adivinanzas.
AlphaFold utiliza un método llamado aprendizaje automático profundo. Para que estas redes neuronales artificiales aprendan se las expone a muchas estructuras de proteínas ya conocidas que corresponden a secuencias de aminoácidos específicas. Una vez entrenado, el sistema es el que debe predecir la forma de secuencias novedosas.
El concepto de aprendizaje automático (o de máquina) se planteó de manera formal en la década de 1950. Sin embargo, como explica el bioquímico mexicano Gabriel del Río Guerra, del Instituto de Fisiología Celular de la unam, por entonces no existían ni algoritmos ni computadoras suficientemente potentes para poner en práctica el concepto. Del Río Guerra cuenta que conforme aumentaron la capacidad y la velocidad de procesamiento de las computadoras fue posible entrenar redes neuronales artificiales para hacer montones de cosas, como reconocer imágenes de casas, flores, personas o trenes (etiquetadas previamente por un humano). Para este entrenamiento se expone a las redes a miles y miles de ejemplos, de modo que puedan encontrar patrones que se correlacionen con las etiquetas. Cuando se hizo lo mismo con la predicción de proteínas, vino el “boom”.
El éxito del aprendizaje de máquina para el reconocimiento de imágenes y otras tareas sugería que era una herramienta adecuada para acometer el problema de la predicción de la estructura proteínica, dice Del Río Guerra. “El ser humano puede hacer predicciones sin entender lo que hace”, continúa. Por ejemplo, los humanos usamos el fuego desde mucho antes de saber nada de termodinámica. Mediante prueba y error pudimos anticipar lo que pasaba cuando se expone un objeto o a una persona al fuego: qué cosas se calientan y cuáles se echan a perder. Y todo esto sin entender el fenómeno en sí. “Eso es exactamente de lo que se trata el aprendizaje de máquina: me das muchos ejemplos y a partir de ellos yo hago una predicción.”
Esto es lo que hizo AlphaFold 2: examinó las aproximadamente 170 000 proteínas cuya estructura ya ha sido determinada por métodos experimentales y aprendió de ellas. “Esto es tremendo”, declaró Moult al enterarse de los resultados que obtuvo el algoritmo en 2020. “En cierto sentido, el problema está resuelto”, dijo.

¿Será?
En un artículo publicado en 2021 Lupas y colegas afirman que el problema se resolvió quizá sólo en su forma básica; es decir, en lo que atañe a la capacidad de deducir la estructura de una proteína a partir de su secuencia de aminoácidos. Pero el problema es más complejo: la secuencia de una proteína contiene también información sobre la que aún no es posible hacer predicciones precisas; por ejemplo, qué moléculas es capaz de reconocer la proteína, a cuáles se adhiere o cómo responde a condiciones cambiantes en el medio. Los autores concluyen que AlphaFold 2 es sólo el primer paso en el camino hacia la solución del problema del plegamiento de proteínas.
Del Río Guerra coincide en que aún hay muchas preguntas pendientes. Predecir la función de la proteína, por ejemplo, sigue siendo un enorme reto, y este problema ha engendrado un concurso equivalente al casp, en el que se ponen a prueba métodos computacionales que predicen la función de las proteínas a partir de su secuencia o estructura. Aunque también aquí se han utilizado métodos de aprendizaje de máquina, los puntajes obtenidos no han sido tan altos como en el casp, explica.
Nina Pastor Colón, investigadora de la Universidad Autónoma del Estado de Morelos, cuenta que existen otras limitaciones de estos algoritmos de inteligencia artificial. Pastor Colón estudia unas moléculas llamadas proteínas intrínsecamente desordenadas o desestructuradas, que no cumplen con el precepto clásico “una secuencia-una estructura”, sino que adoptan diferentes formas dependiendo de las moléculas que las rodean. Por ejemplo, la proteína p53, asociada a la protección contra el cáncer, tiene secciones intrínsecamente desordenadas que usa para unirse a varias proteínas más, y con cada una adopta una configuración distinta. Es “un camaleón molecular”, en palabras de Pastor Colón. Estas secuencias no son raras (hay estimaciones que sugieren que 30 % de las proteínas humanas son intrínsecamente desordenadas), pero su naturaleza las hace difíciles de estudiar a nivel experimental, por lo que los algoritmos de aprendizaje automático no han sido entrenados con estos ejemplos y están muy limitados en el tipo de predicciones que hacen para ellas. Pastor Colón considera que herramientas como AlphaFold pueden ser rápidas, pero no son infalibles.
Sin embargo, como sugiere Lupas, es probable que estos métodos sean sólo el principio de la solución al problema del plegamiento. En diciembre de 2022 se llevó a cabo el más reciente casp. DeepMind y su AlphaFold no participaron esta vez, pero entre los grupos ganadores de ese año la mayoría incorporó el algoritmo de AlphaFold —que es público— y se lograron algunos pequeños avances en los retos pendientes. El año pasado también entró en escena la compañía Meta (antes Facebook), el gigante de las redes sociales, que usando un algoritmo también basado en aprendizaje automático logró predecir la forma de más de 600 millones de proteínas a partir de secuencias derivadas de adn de muestras de suelo y agua de mar, así como de intestino y piel humanos. Lo hizo en apenas dos semanas, más rápido que AlphaFold 2.
El interés por resolver el plegamiento de proteínas no nace sólo de la simple curiosidad. Se discute el alcance que tendrá predecir una estructura sin necesidad de invertir todo el tiempo y el dinero que implica la experimentación, y ahorrándose, por ejemplo, los años y los kilos de carne de cachalote que a Kendrew le exigió su misión. Dada la estrecha relación que hay entre las formas de una proteína y muchos padecimientos —desde el cáncer, el Alzheimer y el Parkinson hasta enfermedades infecciosas como el Chagas o las causadas por rotavirus— poder determinar en minutos una estructura a partir únicamente de la secuencia de aminoácidos podría ayudar a entender estos padecimientos o acelerar el diseño y desarrollo de fármacos para combatirlos.
Ya hay grupos de investigación que usan AlphaFold para desarrollar vacunas más efectivas contra la malaria. Otros más emplean el programa para estudiar proteínas que participan en la resistencia a antibióticos desarrollada por muchas bacterias —un grave problema de salud pública—. Algunos investigadores también están empleando esta tecnología de predicción para diseñar proteínas que aceleren el reciclaje de plásticos de un solo uso.
Sin embargo, también debe quedar claro que predecir la estructura y entender el plegamiento son dos cosas muy diferentes, dice Pastor Colón. “El problema [...] de la inteligencia artificial es que es una bonita caja negra”, explica. Puede ofrecer buenos resultados, pero no sabemos por qué: no conocemos las variables que usa el algoritmo para hacer sus predicciones. “Por lo tanto, uno renuncia a entender cómo le hace para plegarse y se queda con el resultado final del proceso, nada más.”
Esta búsqueda, que requiere mucho más que inteligencia artificial, es algo que motiva a Pastor Colón a seguir estudiando la dinámica molecular de las proteínas. Como concluye la investigadora: no debemos renunciar “al fino arte de pensar”.
- “Funciones de las proteínas”, Portal Académico cch, unam, en: https://e1.portalacademico.cch.unam.mx/alumno/biologia1/unidad1/biomoleculas/funcionesproteinas.
- "Proteínas", Coordinación de Universidad Abierta, Innovación Educativa y Educación a Distancia, unam, en: http://uapas2.bunam.unam.mx/ciencias/proteinas/
Alejandra Manjarrez estudió biología en la unam y es doctora en ciencias por la Escuela Politécnica Federal de Zúrich. Es escritora de ciencia y su trabajo ha sido publicado en The Scientist, The Atlantic, Muy Interesante y Revista de la Universidad de México, entre otros medios.










